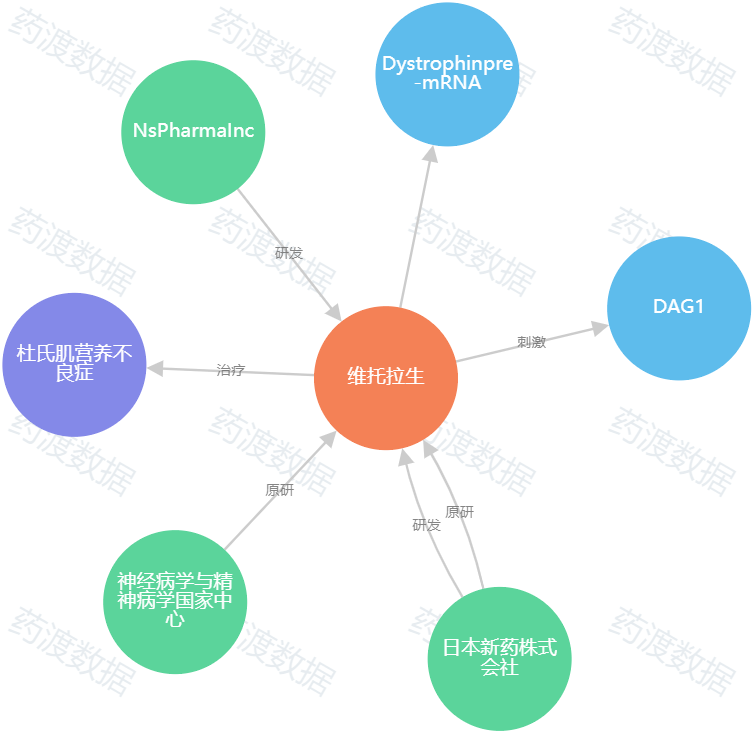
圖1.Viltolarsen知識圖譜(圖源:藥渡數據)
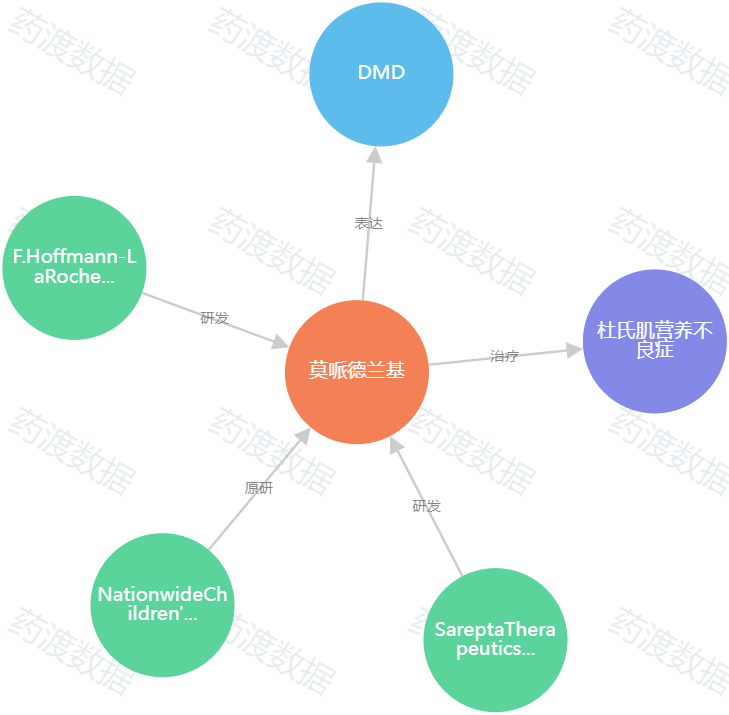
圖2.Elevidys知識圖譜(圖源:藥渡數據)
參考文獻:



合作咨詢
![]() 肖女士
肖女士
![]() 021-33392297
021-33392297
![]() Kelly.Xiao@imsinoexpo.com
Kelly.Xiao@imsinoexpo.com
 2006-2025 上海博華國際展覽有限公司版權所有(保留一切權利)
滬ICP備05034851號-57
2006-2025 上海博華國際展覽有限公司版權所有(保留一切權利)
滬ICP備05034851號-57






















